")
")
Irkutsk scientists using computational population genetics methods have restored the chronology of the first wave of the coronavirus pandemic (SARS-CoV-2)
Specialists from Laboratory of Genosystematics at Limnological Institute, Laboratory of Natural Focal Infectious Diseases at Irkutsk Antiplaque Research Institute of Siberia and the Far East of the Russian Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing (Rospotrebnadzor) and the Research Institute of Bio-Medical Technologies of Irkutsk State Medical University, using the computing resources of Irkutsk Supercomputer Center SB RAS, have reconstructed the history of evolution and spread of the SARS-CoV-2 virus in Russia and the world. The study used information from the GISAID global database. The analysis involved the data on 319 whole viral genomes from Russia and 232 whole genomes from other 20 countries of the world, including China.
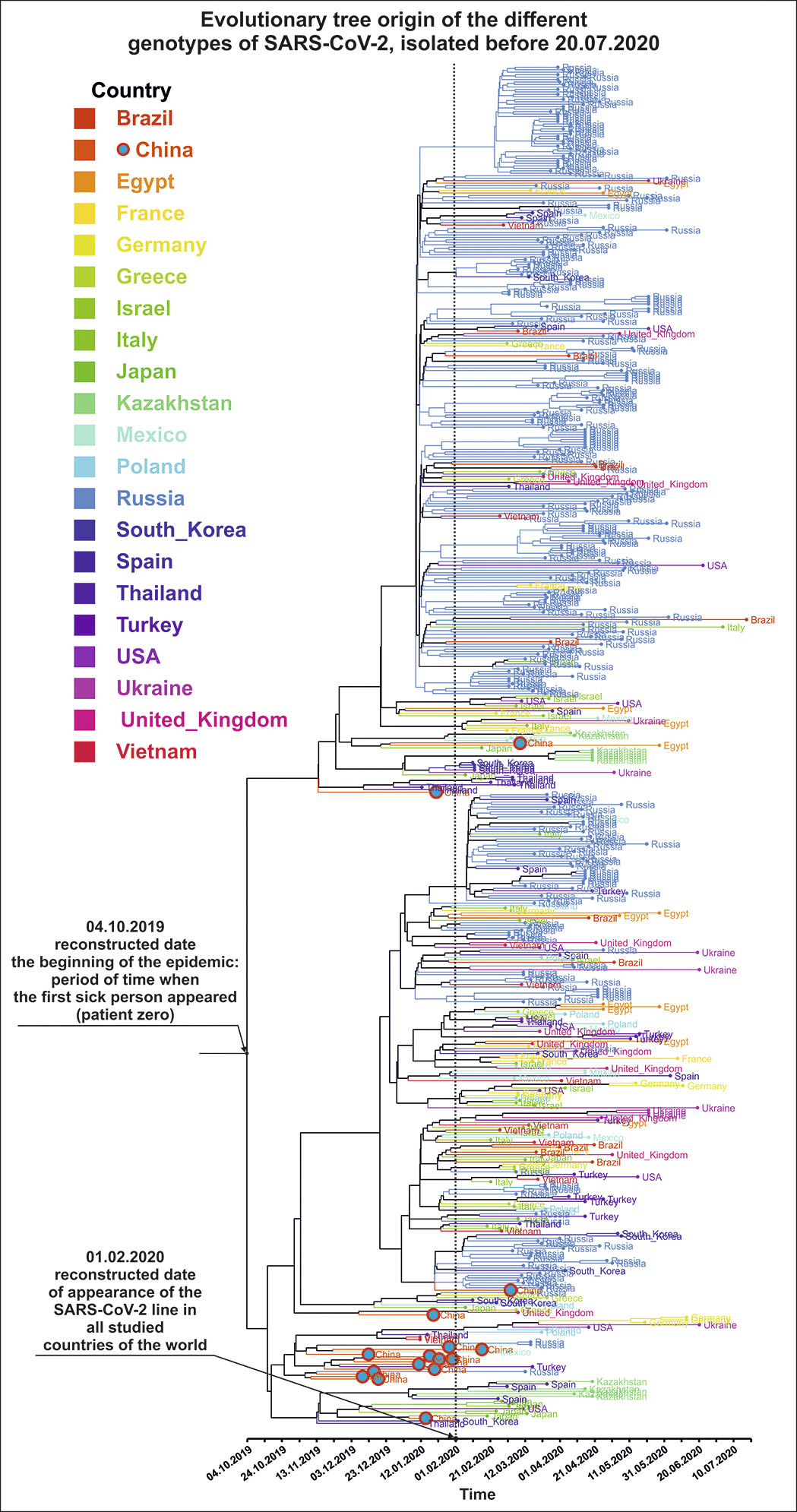
Comparison of viral genomes revealed that the first incidence of the novel SARS-CoV-2 coronavirus in the world occurred from September to November 2019 in China. After that, the genotype of this virus quickly spread almost throughout the world. From mid- to late January 2020, the virus spread to all these 20 countries of Asia, Africa, North and Latin America, and Russia as well as neighbouring countries (see Fig. 1). By the end of February 2020, many sites of the COVID-19 disease had formed in the world and its genetic diversity reached the maximum values (see Fig. 2).
Our study indicates that neither the World Health Organization nor the governments of different countries of the world could control the pandemic and the spread of the disease at the onset of the COVID-19 pandemic. Obviously (see Fig. 2), all especially important decisions to combat the pandemic in all countries of the world were made afterwards, with a delay of one and a half to two months. Even the World Health Organization announced the thread of the worldwide COVID-19 pandemic only on 30 January 2020 when the pandemic was in full swing (see Figs 1 and 2).
|
Fig.1. A reconstructed phylogenetic tree with the molecular clock of the origin of various genotypes of the SARS-CoV-2 virus in Russia and in the world in the initial period of the pandemic. |
In Russia, there are two distinct periods of growth in SARS-CoV-2 effective population size (see Fig. 2). The first period corresponds to the global growth trend from early February to early March 2020. The second period was observed from early to mid-April 2020. We assume that the second period of growth is associated with the introduction of new genotypes in Russians who returned en mass from abroad during this period. Quarantine measures to curb the penetration of the virus from the outside by keeping these people in self-isolation and observation places worked only partially. The effective population size is a value that is directly proportional to the number of viral genotypes circulating in the population (genetic diversity).
This study has revealed that the rate of evolution of SARS-CoV-2 averaging 0.054% of the changes in the genome for one year is lower than the rate of evolution of such viruses as Zika (0.072%), hepatitis C (0.095%), human immunodeficiency virus type 1 (0.135%), measles (0.062%), and Ebola hemorrhagic fever (0.132%) but higher than influenza A viruses (0.0014%). At such rate of evolution, the coronavirus genome will accumulate on average only 1.4 substitution mutations per month. Taking into account the genomic analysis, based on which the virus spread throughout the world virtually in one month, whereas, according to the WHO official data, it took two and a half months, it is not possible to determine the migration pathways and transition of the virus from one country to another only based on genomic data for this period without additional information about the movements of specific sick people.
If the developed vaccines show their effectiveness to prevent the infection and/or development of severe forms of the disease they won’t require a frequent renewal of the antigenic composition. This is a positive consequence of rather a slow rate of accumulation of substitutions in the SARS-CoV-2 genome.
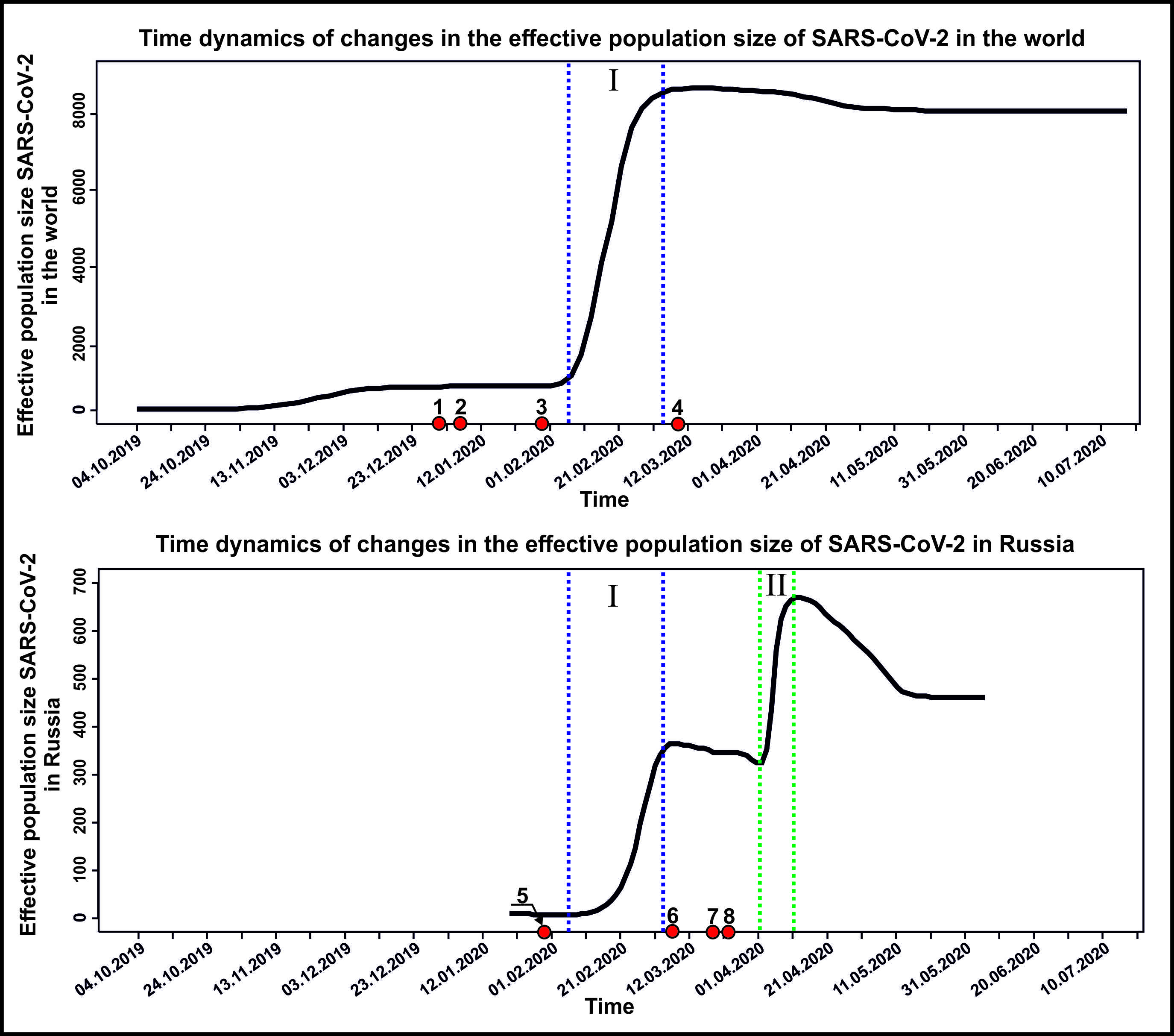 |
Fig.2. Dynamics of changes in the effective population size of SARS-CoV-2 in Russia and in the world in the initial period of the pandemic. I) the first period of rapid growth of the effective population size in Russia and in the world, II) the second period of rapid growth of the effective population size in Russia. The numbers on the time axis indicate the key events in the development of the pandemic: 1- 31.12.2019 China first reported an outbreak of a new, unknown viral infection; 2 – 01.05.2020 in China, the decrypted first genome of SARS-CoV-2, the biological nature of the disease has been established; 3 - 30.01.2020 WHO declared the outbreak of COVID-19 a global health emergency; 4 - 11.03.2020 WHO recognized that the spread of COVID-19 has become a global pandemic; 5 - 31.01.2020 the first two confirmed clinical cases of the COVID-19 in Russia; 6 - 7.03.2020 4 new cases of COVID-19 in Russian citizens who returned from Italy (the official date of the start of the epidemic in Russia); 7 - 19.03.2020 in Russia, mass events are canceled and schools and universities are switching to remote operation; 8 - 26.03.2020 in Russia, air traffic with other countries was terminated, with the exception of charter flights for the return of Russians. |